endovasculares.es




Noticias
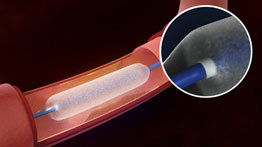
- Título: FDA approves first drug-coated angioplasty balloon catheter to treat vascular disease
- Fecha: 10-10-2014
The U.S.Food and Drug Administration today approved the Lutonix 035 Drug CoatedBalloon Percutaneous Transluminal Angioplasty Catheter (Lutonix DCB). This isthe first drug-coated balloon used to re-open arteries in the thigh(superficial femoral arteries) and knee (popliteal arteries) when narrowed orblocked as a result of peripheral artery disease (PAD).
PAD occurswhen fatty material (plaque) builds up in the arteries that carry blood to thelimbs, usually affecting the arteries in the legs. This causes hardening and/ornarrowing of the arteries (atherosclerosis), limiting the flow of oxygen-richblood to the body. People with PAD may experience symptoms, such as leg pain,or serious complications, including skin ulcers or gangrene. Existing optionsfor treatment of PAD can include exercise, drug therapy, and other optionswithin the artery, such as non-drug coated PTA, bare-metal or drug-elutingstenting, or surgical bypass.
The LutonixDCB is a percutaneous transluminal angioplasty (PTA) catheter. The product hasa balloon that is used to re-open the artery. The balloon is coated on itsouter surface with the drug paclitaxel, which may help to prevent recurrentnarrowing of arteries (restenosis) after the procedure. During the procedure,the artery is first partially opened with a traditional angioplasty balloon,without a drug coating. The Lutonix DCB is then used to fully open the narrowedportion of the artery and apply the drug to the artery wall. The Lutonix DCBmay be used in arteries located in the thigh or the knee.
“Peripheralartery disease can be quite serious. Preventing further blockage of arteries isjust as important as removing the initial blockage” said William Maisel, M.D.,M.P.H., deputy director for science and chief scientist in the FDA’s Center forDevices and Radiological Health. “The clinical data show that Lutonix DCB maybe more effective than traditional balloon angioplasty at helping to preventfurther blockage in the artery.”
Thedemonstration of safety and effectiveness of the Lutonix DCB came fromnonclinical testing, as well as three clinical studies.
Onerandomized, multi-center, European clinical study compared the safety andeffectiveness of the Lutonix DCB to conventional balloon angioplasty. The studyenrolled 101 participants who were randomly selected to be treated with LutonixDCB or conventional balloon angioplasty. At the end of six months, 71.8 percentof participants treated with Lutonix DCB did not require additional PADtreatment compared to 48.6 percent of those treated with conventional balloonangioplasty.
In aseparate pivotal, single blind, multi-center study conducted in the UnitedStates and Europe, researchers enrolled 476 participants who were randomlyselected to be treated with Lutonix DCB or conventional balloon angioplasty. Atthe end of 12 months, 65.2 percent of participants treated with Lutonix DCB didnot have a narrowing of the arteries (restenosis) compared to 52.6 percent ofthe control group.
Asingle-arm safety study involving the Lutonix DCB was also initiated and isongoing. Researchers enrolled 657 participants in the United States and Europe.The primary objective of this safety study is to collect additional safety andeffectiveness data on the Lutonix DCB in a large population. The availableresults at the time of product approval show that there have been nounanticipated device- or drug-related adverse events.
Thesestudies also indicated that the safety of Lutonix DCB was comparable toconventional balloon angioplasty. The most common major adverse events includedadditional intervention, pain as a result of poor blood flow, narrowing ofarteries that were not treated, chest pain, and abnormal growth oftissue.
The productis contraindicated (should not be used) in patients who cannot receiverecommended drug therapy due to bleeding disorders; patients who cannot take orhave known hypersensitivities to paclitaxel or structurally-related compounds;women who are breastfeeding, pregnant, or plan to become pregnant; or menintending to father children.
As part ofthe approval, the FDA is requiring the manufacturer to conduct two post-approvalstudies. One is a five-year post-approval study of 657 patients treated withthe Lutonix DCB to further monitor safety and effectiveness. The second is arandomized, single blind, multi-center study which will assess the safety andeffectiveness of the Lutonix DCB in women in the United States, due todifferences in observed outcomes in this group as compared to outcomes for thegeneral study population.
Lutonix DCBis manufactured by Lutonix, Inc. of New Hope, Minnesota.
The FDA, anagency within the U.S. Department of Health and Human Services, protects thepublic health by assuring the safety, effectiveness, and security of human andveterinary drugs, vaccines and other biological products for human use, andmedical devices. The agency also is responsible for the safety and security ofour nation’s food supply, cosmetics, dietary supplements, products that giveoff electronic radiation, and for regulating tobacco products.
- Fuente: endovascular.es
@2011 endovascular. Última actualización: 31-03-2014
